Keyora Nutritional Neurology – Ashwagandha · Episode 1

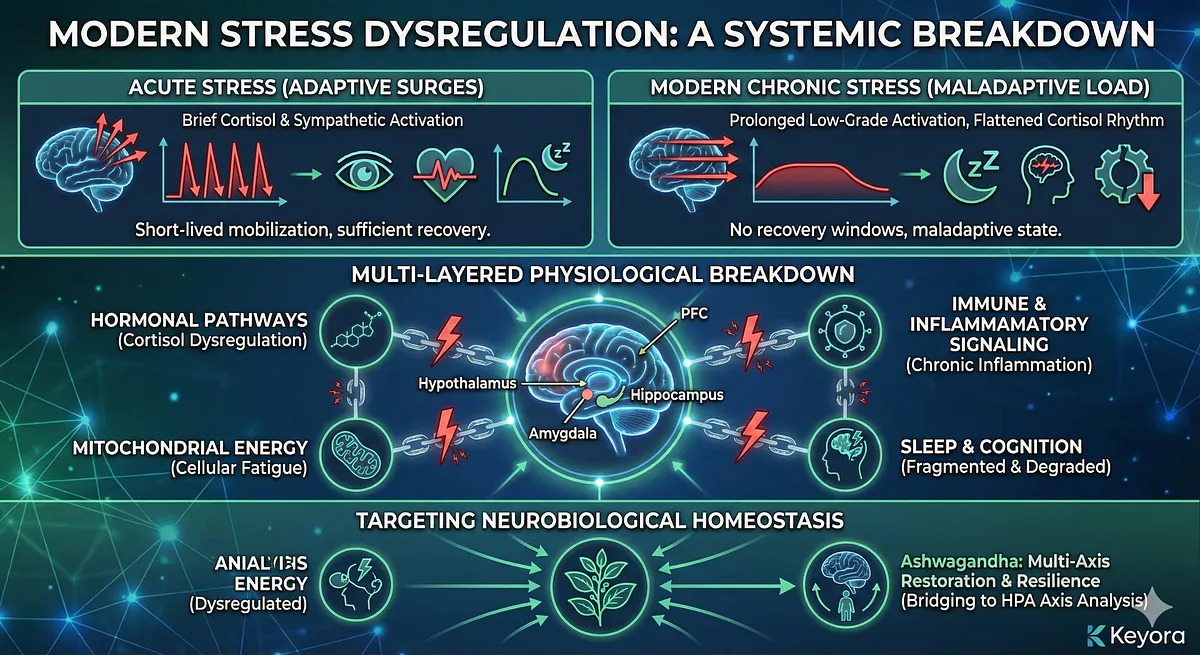
Modern stress is no longer defined by brief, adaptive surges of alertness.
Instead, it presents as a persistent and dysregulated biological load that disrupts the entire neuro-endocrine-inflammatory network.
Unlike acute stress – which relies on a short-lived mobilization of cortisol, sympathetic activation, and heightened cognitive focus – modern individuals experience prolonged, low-grade activation of the same systems without sufficient recovery windows.
This chronic activation forces the stress machinery of the brain and body into a maladaptive state, flattening cortisol rhythms, fragmenting sleep, destabilizing emotional circuits, and degrading cognitive performance.
These changes are not psychological in origin; they emerge from the systems biology of stress dysregulation. The brain’s stress circuitry – spanning the hypothalamus, amygdala, hippocampus, and prefrontal cortex - interacts continuously with hormonal pathways, immune responses, inflammatory signaling, and mitochondrial energy systems.
When stress becomes chronic, these interconnected mechanisms lose synchrony, resulting in a multi-layered breakdown of physiological resilience.
This chapter establishes the scientific foundation required before examining the HPA axis in detail.
By understanding how modern stress interacts with sleep, emotion, cognition, inflammation, and cellular energy, we can clarify why interventions such as Ashwagandha – targeting multiple regulatory axes simultaneously – have become essential tools in restoring neurobiological homeostasis.
The following sections outline this collapse step by step, forming the conceptual bridge between the Preface and the mechanistic analysis in Episode 2.
– Modern stress differs from acute stress: chronic, persistent, low-grade activation.
– Disrupts entire neuro-endocrine-inflammatory network.
– Mechanisms: flattened cortisol rhythm, sleep fragmentation, emotional instability, cognitive decline.
– Involves multi-system failure: HPA axis, limbic system, immune signaling, mitochondrial energy.
– Purpose of Episode 1: establish systems biology framework before Episode 2 (HPA axis).
By Keyora Research Notes Series
This article contributes to Keyora’s ongoing scientific documentation series, which systematically outlines the conceptual foundations, mechanistic pathways, and empirical evidence informing our research and development approach.
ORCID: 0009–0007–5798–1996
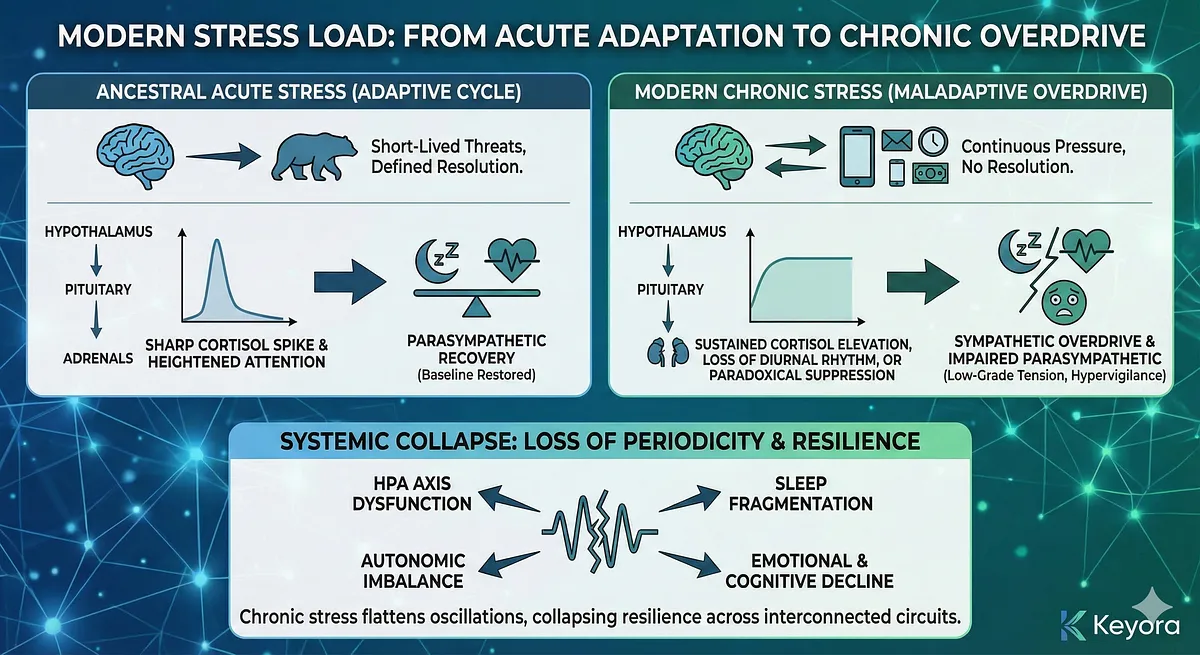
Section I – Modern Stress Load:
From Acute Adaptation to Chronic Overdrive
In ancestral environments, the stress response was a survival mechanism – rapid, targeted, and short-lived.
Activation of the amygdala initiated an immediate cascade through the hypothalamus and sympathetic nervous system, triggering cortisol release, heightened attention, and increased metabolic readiness. Once the threat passed, parasympathetic recovery restored baseline physiology.
This clean, cyclical pattern enabled humans to maintain health while responding to intermittent danger.
Modern life has dissolved this cycle.
Stressors now present not as acute threats but as continuous, psychologically mediated pressures: digital overload, constant notifications, unstable work rhythms, emotional labor, competitive academic environments, financial uncertainty, and the blurring of rest-work boundaries.
These inputs activate the same ancient biological circuitry, but without defined resolution points. As a result, the brain’s stress machinery becomes locked in a semi-activated state, neither fully “on” nor capable of returning “off.”
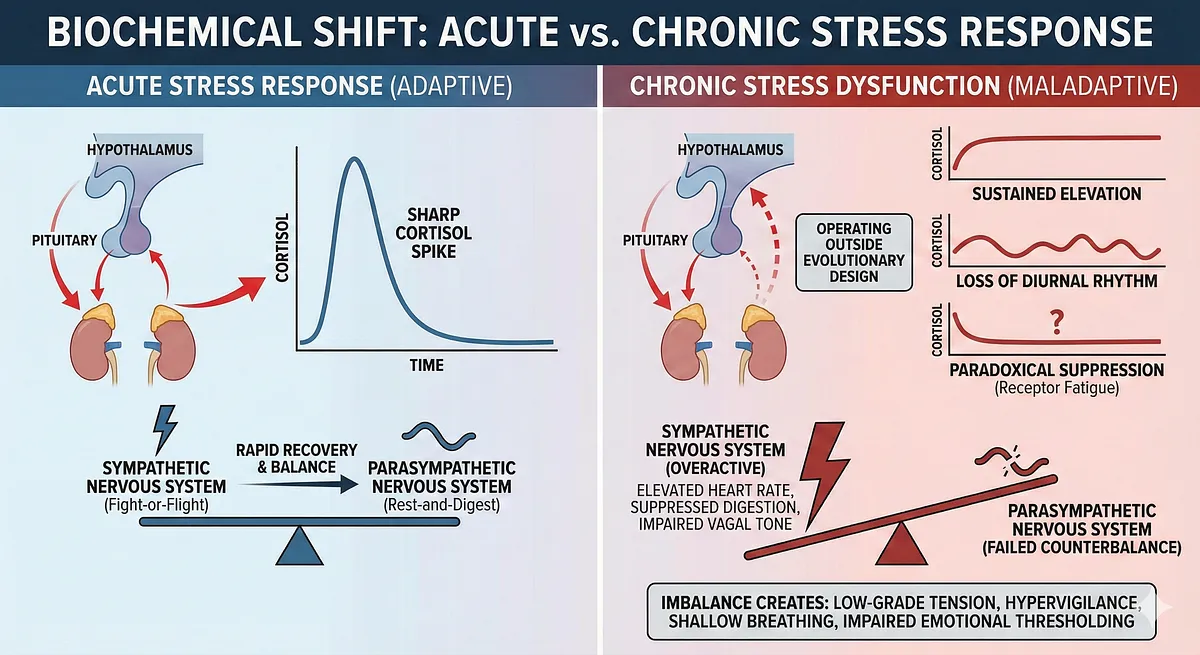
At the biochemical level, this shift forces the HPA axis to operate outside its evolutionary design. Instead of generating a sharp cortisol spike followed by decline, chronic stress produces:
- sustained cortisol elevation,
- loss of diurnal rhythm, or
- paradoxical cortisol suppression caused by receptor fatigue and feedback failure.
Any of these patterns indicate that the acute-phase machinery has transitioned into a chronic-phase dysfunction.
Simultaneously, the sympathetic nervous system remains overactive – elevating heart rate, suppressing digestion, and impairing vagal tone – while the parasympathetic system fails to counterbalance.
This imbalance creates a biological environment dominated by low-grade tension, hypervigilance, shallow breathing, and impaired emotional thresholding.
From a systems-level perspective, the problem is not simply “more stress,” but the loss of periodicity and recovery. Biological systems rely on oscillation – peak and trough – to maintain homeostasis.
Chronic stress flattens these oscillations, collapsing resilience across multiple interconnected circuits. This collapse is the starting point for understanding why the stress–sleep–emotion–cognition network fails and why HPA axis dysfunction becomes inevitable.
– Acute stress = short-term, adaptive, high-resolution cycle; modern stress = continuous and unresolved.
– Chronic psychological and environmental stressors activate ancient circuitry without recovery.
– Leads to sustained cortisol elevation, loss of rhythm, or paradoxical suppression.
– Sympathetic overactivation + poor parasympathetic recovery → chronic tension and hypervigilance.
– Core problem: loss of biological periodicity → collapse of resilience across systems.
Section II
Disrupted Cortisol Rhythmicity as the Earliest Signal of Stress-System Failure
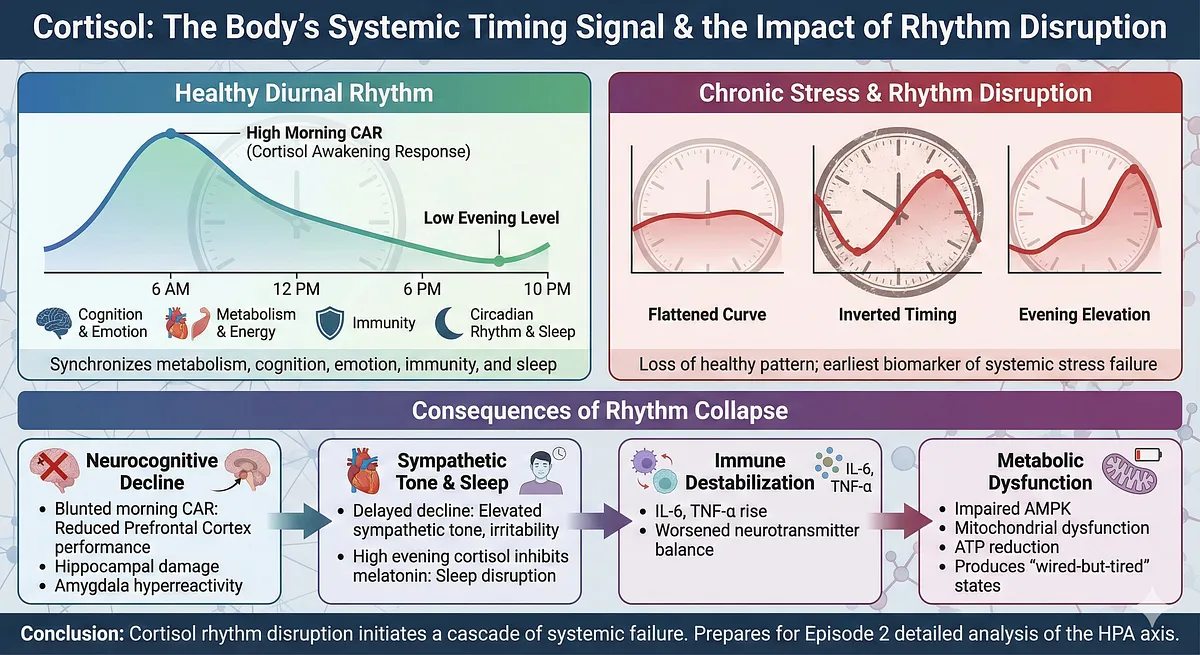
Cortisol is often misunderstood as merely a “stress hormone,” but biologically, it functions as the master timing signal that synchronizes metabolic activity, cognitive alertness, emotional stability, immune function, and circadian systems across the brain and body.
Under healthy conditions, cortisol follows a precise diurnal pattern: a sharp morning peak known as the cortisol awakening response (CAR), a gradual decline across the day, and a stable low during the evening to allow melatonin release and sleep initiation.
This rhythm acts as the central pacemaker for physiological performance.
Chronic stress disrupts this architecture long before overt symptoms emerge. Instead of a well-defined morning peak, individuals may experience a flattened curve, inverted timing, or a low morning/high evening drift.
Each pattern reflects a different stage of HPA axis dysregulation, indicating that the system has shifted from adaptive pulsatility to maladaptive tonic activation.
Flattening suggests chronic overdrive and receptor desensitization; inversion indicates impaired hypothalamic signaling; and evening elevations are strongly associated with anxiety circuits, emotional lability, and sleep fragmentation.
Disruption in cortisol rhythmicity produces cascading consequences. A blunted morning CAR reduces prefrontal activation, impairing planning, attention, and emotional regulation.
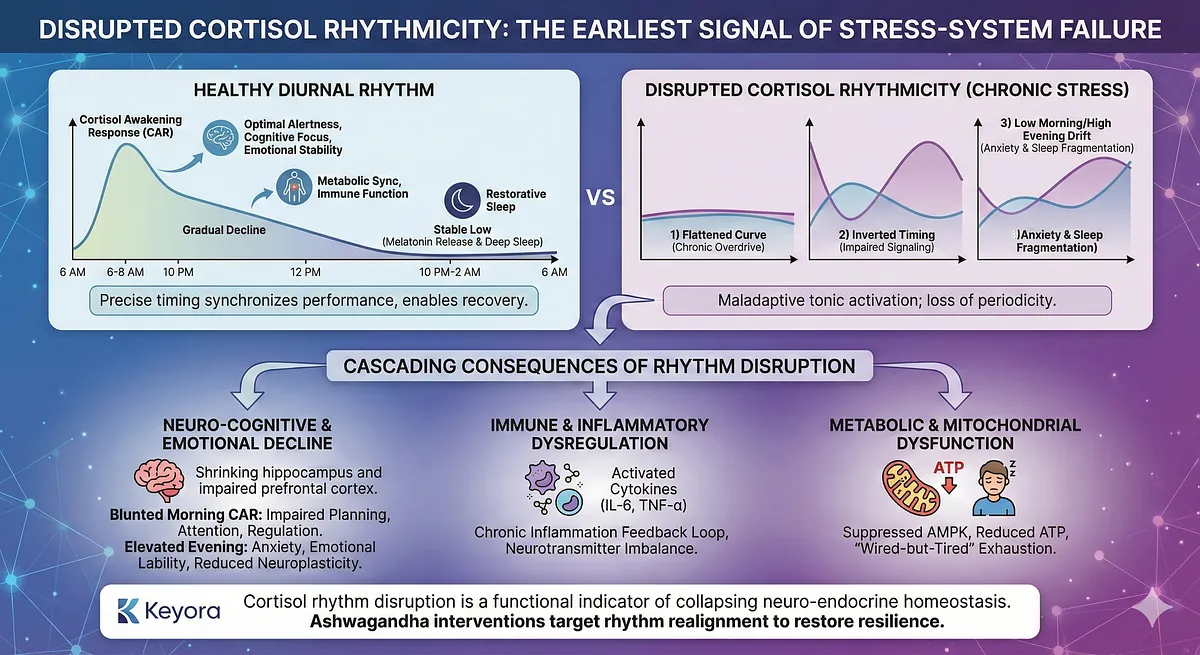
A delayed decline keeps sympathetic tone elevated through the afternoon, leading to irritability, sensory hypersensitivity, and cognitive fatigue. Elevated evening cortisol inhibits melatonin release, reducing sleep depth and shortening slow-wave phases, which are essential for memory consolidation and neuroplasticity.
Over time, this dysregulation contributes to hippocampal shrinkage, impaired amygdala gating, and a reduced threshold for emotional reactivity.
Importantly, altered cortisol rhythms also reprogram immune and inflammatory networks.
A healthy circadian cortisol signal suppresses excessive inflammatory cytokines. When the rhythm collapses, immune cells shift into a chronically activated state, producing IL-6, TNF-α, and other mediators that further disrupt neurotransmitter balance.
The result is a feedback loop: inflammation worsens cortisol regulation, and cortisol dysregulation fuels further inflammation.
This reciprocal destabilization is a defining characteristic of modern stress physiology.
Disrupted cortisol rhythmicity also has metabolic and mitochondrial implications. Cortisol’s timing influences glucose availability, insulin sensitivity, and mitochondrial biogenesis.
Chronically elevated or mistimed cortisol suppresses AMPK signaling, reduces ATP generation efficiency, and impairs mitochondrial dynamics in neurons.
This contributes to the “wired-but-tired” presentation – exhaustion coexisting with hyperarousal-now common in high-demand cognitive environments.
Together, these mechanisms show that cortisol rhythm disruption is not merely a biomarker; it is the earliest functional indicator of a collapsing neuro-endocrine homeostasis.
Recognizing this pattern provides essential context for understanding how interventions such as Ashwagandha realign the HPA axis, restore oscillatory patterns, and re-establish the biological periodicity required for resilience.
This sets the stage for Episode 2, where the HPA axis will be dissected in full mechanistic detail.
– Cortisol is a systemic timing signal regulating metabolism, cognition, emotion, immunity, and circadian rhythm.
– Healthy rhythmicity: high morning peak (CAR) → steady decline → low evening level.
– Chronic stress produces flattened curves, inverted timing, or evening elevations.
– Consequences:
– Reduced prefrontal cortex performance when morning CAR is blunted.
– Elevated sympathetic tone and irritability when decline is delayed.
– Sleep disruption from high evening cortisol inhibiting melatonin.
– Hippocampal damage and amygdala hyperreactivity.
– Cortisol rhythm collapse destabilizes immune function: IL-6, TNF-α rise → worsened neurotransmitter balance.
– Metabolic effects: impaired AMPK, mitochondrial dysfunction, ATP reduction → “wired-but-tired” states.
– Rhythm disruption is the earliest biomarker of systemic stress failure.
– Prepares for Episode 2 detailed analysis of the HPA axis.
Section III
Neuro-Inflammatory Coupling: How Chronic Stress Rewires Immune Signaling
Chronic stress activates a profound reconfiguration of the immune system, shifting it from an adaptive surveillance state into a persistent pro-inflammatory mode that fundamentally alters neural function.
Under healthy conditions, brief cortisol pulses suppress excessive cytokine activity, preventing unchecked inflammation and maintaining stable neurotransmission. However, when stress becomes chronic and cortisol rhythmicity collapses, this anti-inflammatory gating mechanism fails.
Immune cells-particularly microglia, monocytes, and peripheral macrophages – enter a state of heightened reactivity, characterized by increased synthesis of IL-6, TNF-α, IL-1β, and other inflammatory mediators.
These cytokines, once beneficial during acute threats, now become long-acting disruptors of neural signaling and synaptic communication.
The amygdala–hypothalamus–brainstem stress axis plays a central role in this shift. Amygdalar hyperactivity amplifies sympathetic output, which directly stimulates bone marrow and spleen activity, increasing the trafficking of inflammatory monocytes into circulation.
Meanwhile, vagal tone is suppressed, weakening the cholinergic anti-inflammatory reflex that normally terminates cytokine release.
This dual effect-sympathetic overdrive and parasympathetic suppression—creates the perfect environment for systemic inflammation to sustain itself independent of the original stressor.
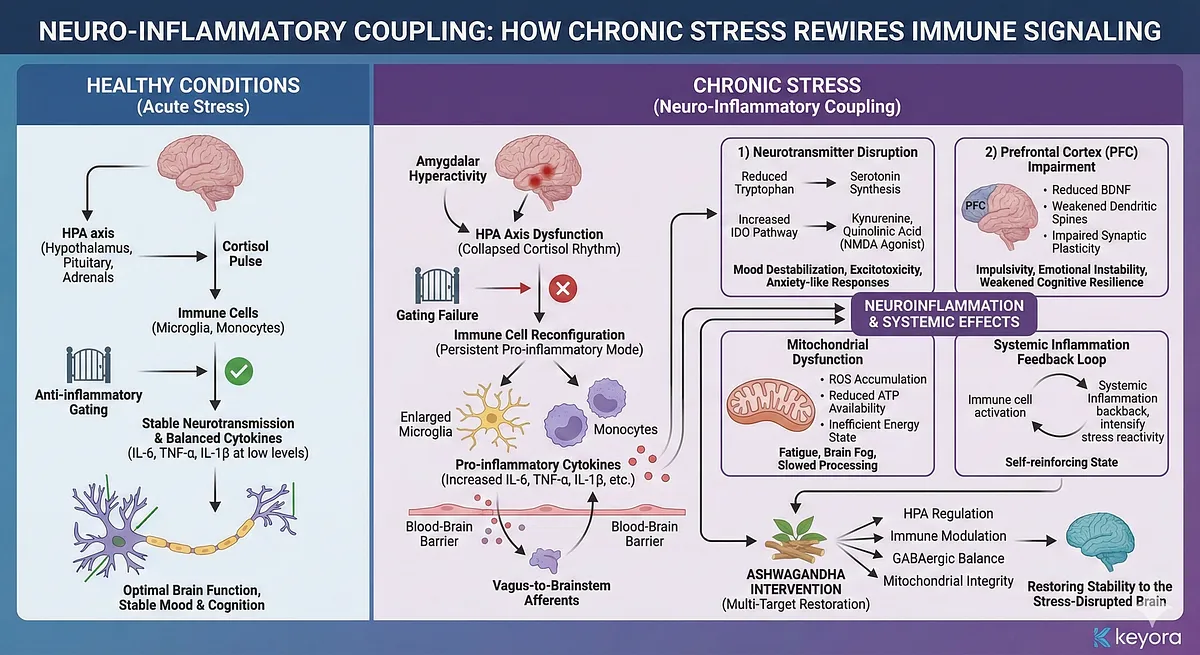
Cytokines exert potent effects on the brain through both humoral and neural pathways. IL-6 and TNF-α cross the blood–brain barrier at specific transport sites, while peripheral inflammation signals the brain via vagus-to-brainstem afferents. Microglia respond by shifting into a “primed” state – morphologically enlarged, metabolically glycolytic, and hyper-responsive to stimuli.
Primed microglia overproduce reactive oxygen species (ROS), quinolinic acid, prostaglandins, and additional cytokines, generating a feed-forward loop of neuroinflammation. This feedback not only intensifies stress reactivity but changes the baseline architecture of neural circuits.
The consequences of this immune reprogramming extend across neurotransmitter systems.
Elevated IL-1β and TNF-α suppress synaptic serotonin release and reduce tryptophan availability by activating the indoleamine 2,3-dioxygenase (IDO) pathway, diverting tryptophan away from serotonin synthesis toward kynurenine metabolites. Some of these metabolites, including quinolinic acid, act as NMDA receptor agonists, increasing excitotoxic load and destabilizing mood and cognition.
At the same time, IL-6 impairs GABAergic inhibitory tone and reduces neurosteroid synthesis, enhancing anxiety-like responses and stress-induced hypervigilance.
Inflammation also disrupts the prefrontal cortex (PFC), the neural hub for executive function and emotional regulation.
Pro-inflammatory cytokines reduce BDNF expression, weaken dendritic spine density, and impair synaptic plasticity in PFC circuits.
This remodeling shifts the functional balance of the limbic–PFC axis: the amygdala becomes hyperactive and overly dominant, while PFC-mediated regulatory control diminishes.
The result is a neurobiological phenotype characterized by impulsivity, emotional instability, rumination, catastrophizing, and weakened cognitive resilience.
Furthermore, the chronic activation of immune–metabolic pathways imposes a substantial burden on mitochondrial function. Cytokine-driven ROS accumulation disrupts mitochondrial membrane potential (ΔΨm), reduces ATP availability, and forces neurons into inefficient energy states.
This metabolic friction further reduces neural stability, aggravating fatigue, brain fog, slowed processing speed, and reduced learning capacity.
In essence, inflammation shifts the brain from a high-efficiency computational state to an energetically compromised mode.
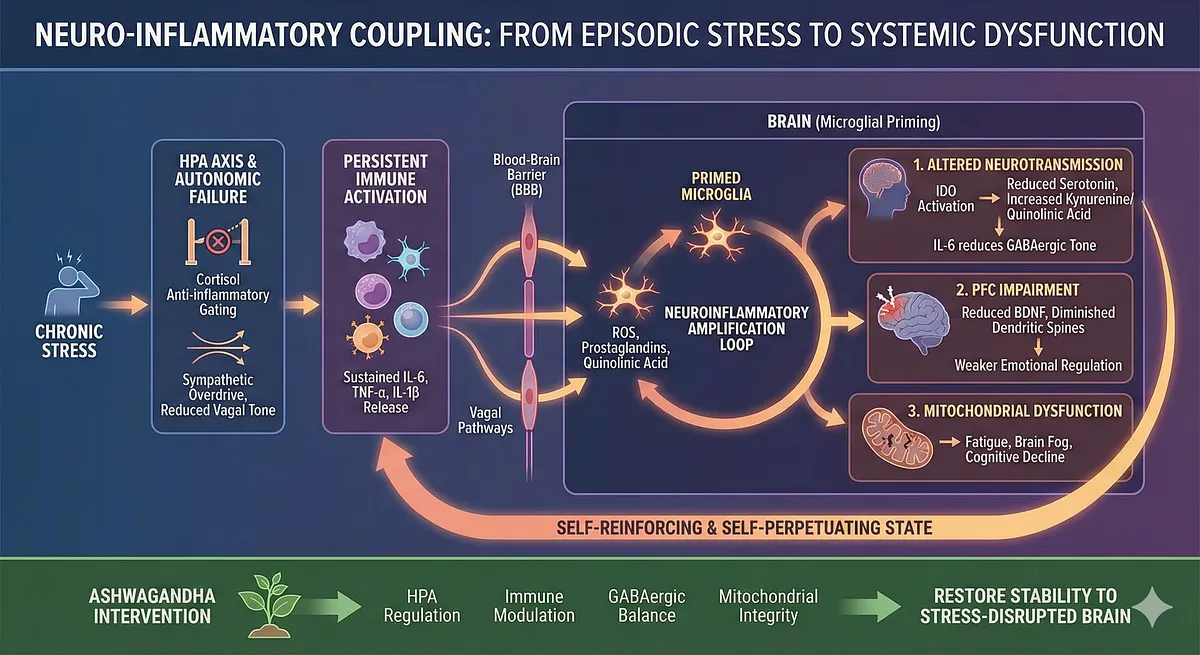
Neuro-inflammatory coupling therefore represents a core mechanism linking chronic stress to long-term psychological, cognitive, and physical dysfunction. It transforms stress from an episodic challenge into a systemic biological state – self-reinforcing and self-perpetuating.
Understanding this mechanism is essential for appreciating why interventions such as Ashwagandha – acting simultaneously on HPA regulation, immune modulation, GABAergic balance, and mitochondrial integrity – are uniquely suited for restoring stability to the stress-disrupted brain.
– Chronic stress collapses cortisol’s anti-inflammatory gating, triggering sustained IL-6, TNF-α, IL-1β release.
– Sympathetic overdrive + reduced vagal tone → persistent immune activation.
– Cytokines affect the brain via BBB transport and vagal pathways → microglial priming.
– Primed microglia produce ROS, prostaglandins, quinolinic acid → neuroinflammatory amplification loop.
– Inflammation alters neurotransmission:
– IDO activation → reduced serotonin, increased kynurenine/quinolinic acid.
– IL-6 reduces GABAergic tone.
– Prefrontal cortex impairment: reduced BDNF, diminished dendritic spines → weaker emotional regulation.
– Cytokine-driven mitochondrial dysfunction → fatigue, brain fog, cognitive decline.
– Neuro-inflammatory coupling converts stress into a self-perpetuating biological state.
Section IV
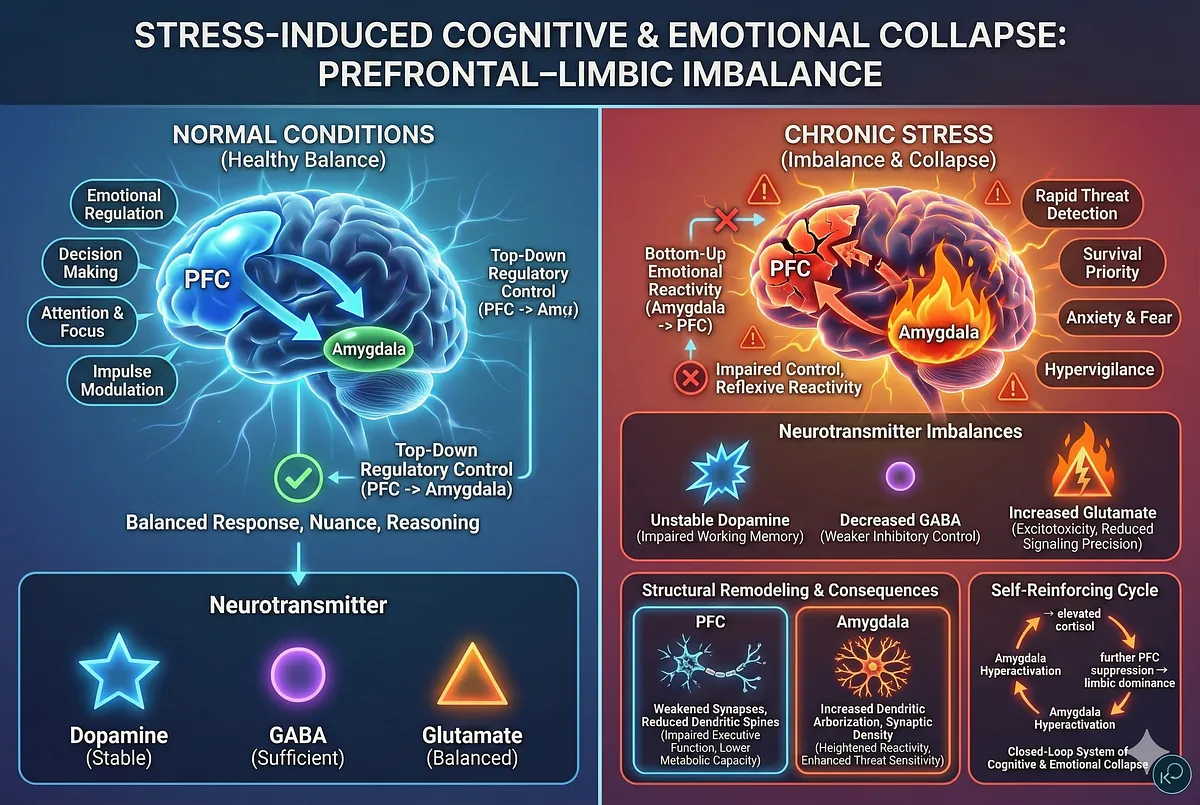
Stress-Induced Cognitive & Emotional Collapse: Prefrontal–Limbic Imbalance
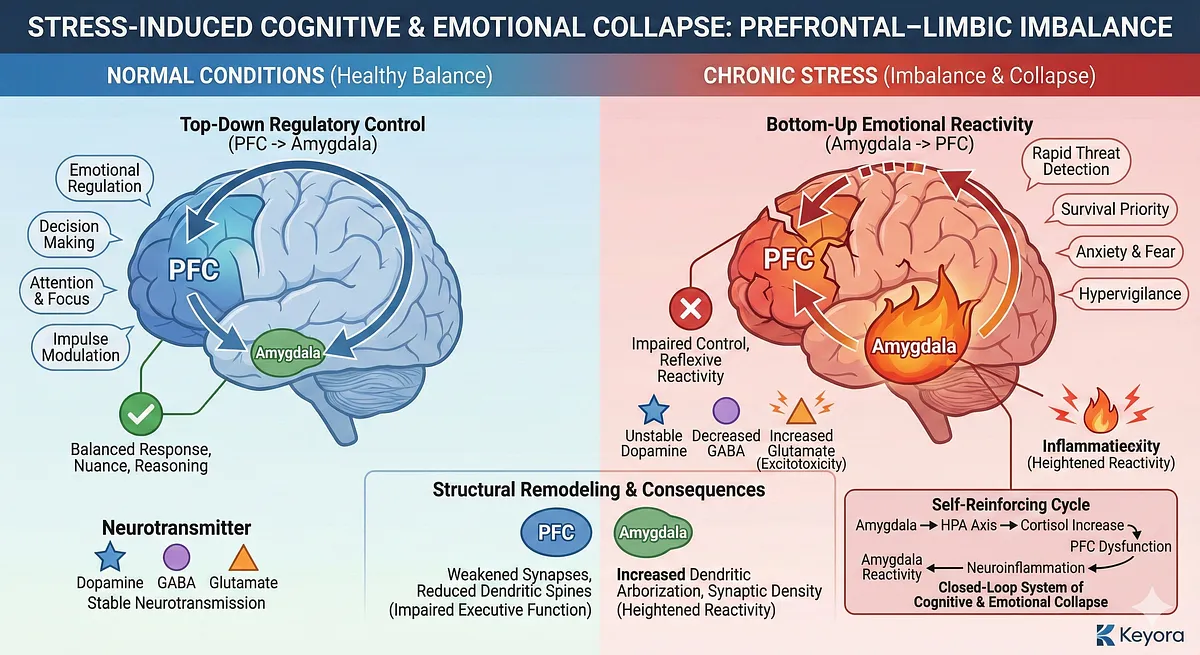
Chronic stress alters the functional architecture of the brain by reshaping the dynamic balance between the prefrontal cortex (PFC) and the limbic system – particularly the amygdala.
Under normal conditions, the PFC exerts top-down regulatory control over emotional responses, threat interpretation, attention, decision-making, and impulse modulation. The amygdala, in contrast, functions as a rapid threat detector, designed to prioritize survival over nuance and long-term reasoning.
These two systems operate in a reciprocal loop: the amygdala signals urgency, and the PFC evaluates context, suppresses unnecessary alarm, and coordinates an adaptive response.
Chronic stress disrupts this regulatory loop at multiple levels. Elevated cortisol, glutamate accumulation, and neuroinflammatory signaling weaken PFC synaptic integrity, reduce dendritic spine density – especially in the medial PFC—and impair neuroplasticity-dependent learning processes.
Over time, this structural remodeling diminishes the PFC’s capacity to maintain executive control. Functional MRI studies consistently show reduced PFC activation during stress, accompanied by hyperactivation in amygdala and subcortical limbic regions.
The result is a shift from reflective, goal-directed processing to reflexive, emotion-driven reactivity.
Mechanistically, chronic stress induces a shift in neurotransmitter balance.
Dopaminergic tone in the PFC becomes unstable, disrupting working memory and attention.
GABAergic inhibition decreases, reducing the brain’s capacity to buffer limbic overactivation.
Simultaneously, excitatory glutamate release increases, promoting excitotoxicity and impairing synaptic precision.
Combined with microglial priming from Section III, this shift in neurotransmission reduces the fidelity of neural signaling across PFC–limbic circuits.
The amygdala undergoes its own form of stress-related remodeling. Unlike the PFC, chronic stress increases dendritic arborization and synaptic density within the basolateral amygdala, making emotional circuits more reactive and less discriminating.
This plasticity bias toward threat sensitivity heightens anxiety, accelerates fear conditioning, and increases susceptibility to emotional contagion. Small stressors trigger disproportionately large responses; ambiguous cues are interpreted as threats; emotional memory traces become more persistent and intrusive.
One of the most immediate functional consequences is impaired attentional gating. The PFC can no longer efficiently filter irrelevant stimuli, leading to hypervigilance, distractibility, and sensory overload.
Cognitive switching slows, decision-making becomes more rigid or impulsive, and working memory capacity drops. These deficits are particularly pronounced in knowledge workers, students, and individuals facing continuous cognitive demands, where PFC-driven executive function is central to daily performance.
Emotionally, the limbic dominance produces instability characterized by irritability, low frustration tolerance, mood reactivity, rumination, catastrophizing, and diminished stress resilience.
The individual becomes more sensitive to social cues, more prone to perceived rejection, and more vulnerable to stress-amplifying internal narratives. This phenotype is not a psychological weakness but the natural neurobiological consequence of losing PFC regulatory authority.
Importantly, this PFC–limbic imbalance becomes self-reinforcing. Heightened amygdala activation further stimulates the HPA axis, elevating cortisol and perpetuating the cycle described in Section II.
Reduced PFC function diminishes the brain’s capacity to downregulate stress responses, while inflammation described in Section III continues to erode synaptic stability.
The result is a closed-loop system of cognitive and emotional collapse—one that cannot be corrected by willpower or lifestyle alone, but requires targeted biological intervention.
Understanding this neural imbalance is crucial for appreciating how adaptogens such as Ashwagandha restore regulatory integrity through modulation of cortisol rhythms, enhancement of GABAergic signaling, suppression of neuroinflammation, and improved mitochondrial efficiency.
The next section will examine how chronic stress parameters ultimately converge on energy metabolism and mitochondrial strain, amplifying the “wired-but-tired” state characteristic of modern stress disorders.
– PFC normally regulates emotion and executive function; amygdala detects threats.
– Chronic stress weakens PFC structure: reduced dendritic spines, impaired plasticity, lower metabolic capacity.
– Amygdala becomes hyperactive: increased excitability, enhanced threat sensitivity, greater emotional reactivity.
– Neurotransmitter imbalances:
– Reduced dopaminergic stability in PFC → impaired working memory.
– Lower GABA → weaker inhibitory control.
– Higher glutamate → excitotoxicity and reduced signaling precision.
– Functional outcomes: distractibility, impaired decision-making, hypervigilance, rumination, emotional instability.
– Self-reinforcing cycle: amygdala hyperactivation → elevated cortisol → further PFC suppression → worsening limbic dominance.
– Sets stage for understanding mitochondrial/energy failure in Section V.
Section V
Mitochondrial Overload, Energy Failure, and the ‘Wired-but-Tired’ Phenotype
Chronic stress imposes a significant metabolic burden on the brain by forcing neural networks to operate under conditions of persistent sympathetic activation, elevated glutamate, disrupted cortisol rhythmicity, and sustained inflammatory signaling.
The brain is the most energy-intensive organ in the human body, relying on precisely regulated mitochondrial function to support synaptic transmission, neurotransmitter cycling, plasticity, and cognitive endurance.
Under prolonged stress, however, mitochondrial systems are pushed into a state of overload, unable to maintain the metabolic demands of hyperarousal while simultaneously supporting cellular repair and restoration.
This mismatch between demand and capacity lies at the core of the “wired-but-tired” phenotype.
Cortisol, when properly rhythmic, enhances mitochondrial biogenesis, ATP production, and metabolic flexibility. But when chronically elevated or mistimed, cortisol becomes mitochondrial-toxic.
It increases oxidative pressure, impairs mitochondrial protein folding, and disrupts electron transport chain (ETC) efficiency – particularly at complexes I and III.
This results in increased leakage of electrons, generating reactive oxygen species (ROS) that further destabilize mitochondrial membranes and reduce membrane potential (ΔΨm). Once ΔΨm decreases below a functional threshold, ATP synthesis becomes insufficient to meet neuronal metabolic demand.
Simultaneously, the inflammatory cascade described in Section III accelerates mitochondrial dysfunction. Cytokines such as TNF-α and IL-1β activate NF-κB and increase inducible nitric oxide synthase (iNOS) activity, producing large amounts of nitric oxide (NO). Excess NO combines with superoxide to form peroxynitrite (ONOO-), one of the most damaging oxidative species for mitochondrial DNA (mtDNA), cardiolipin, and membrane proteins.
Damage to mtDNA reduces the expression of ETC components, creating a downward spiral in ATP availability. Neurons, especially in prefrontal and hippocampal circuits, begin to experience metabolic insufficiency even in the presence of normal oxygen and glucose supply.
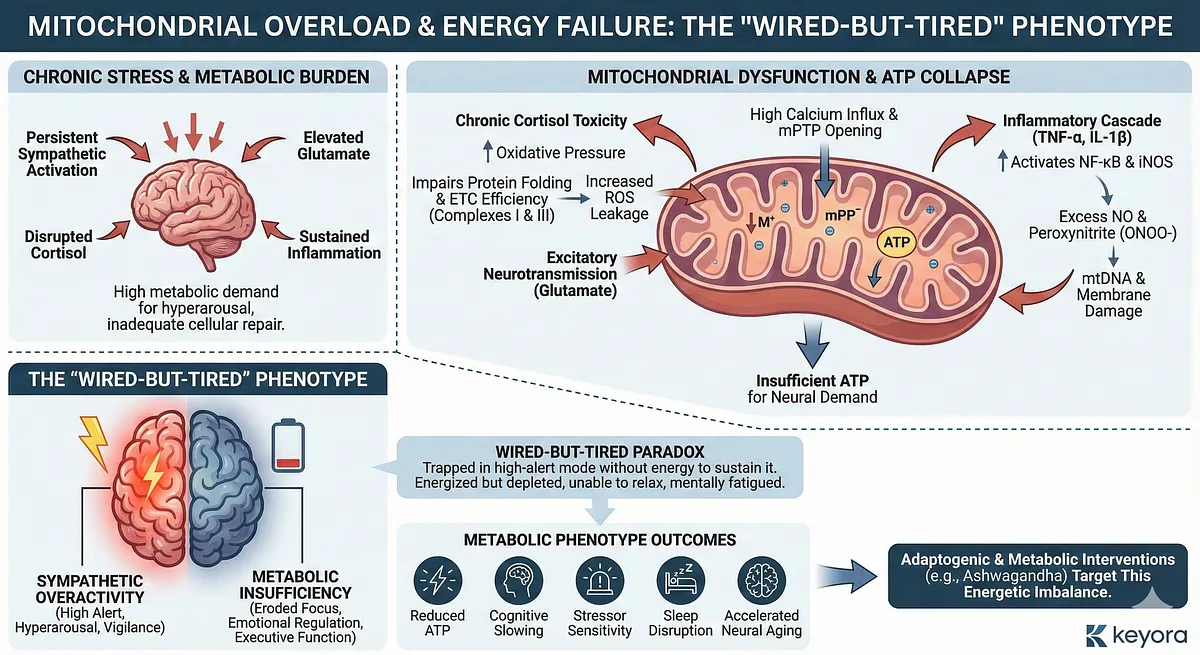
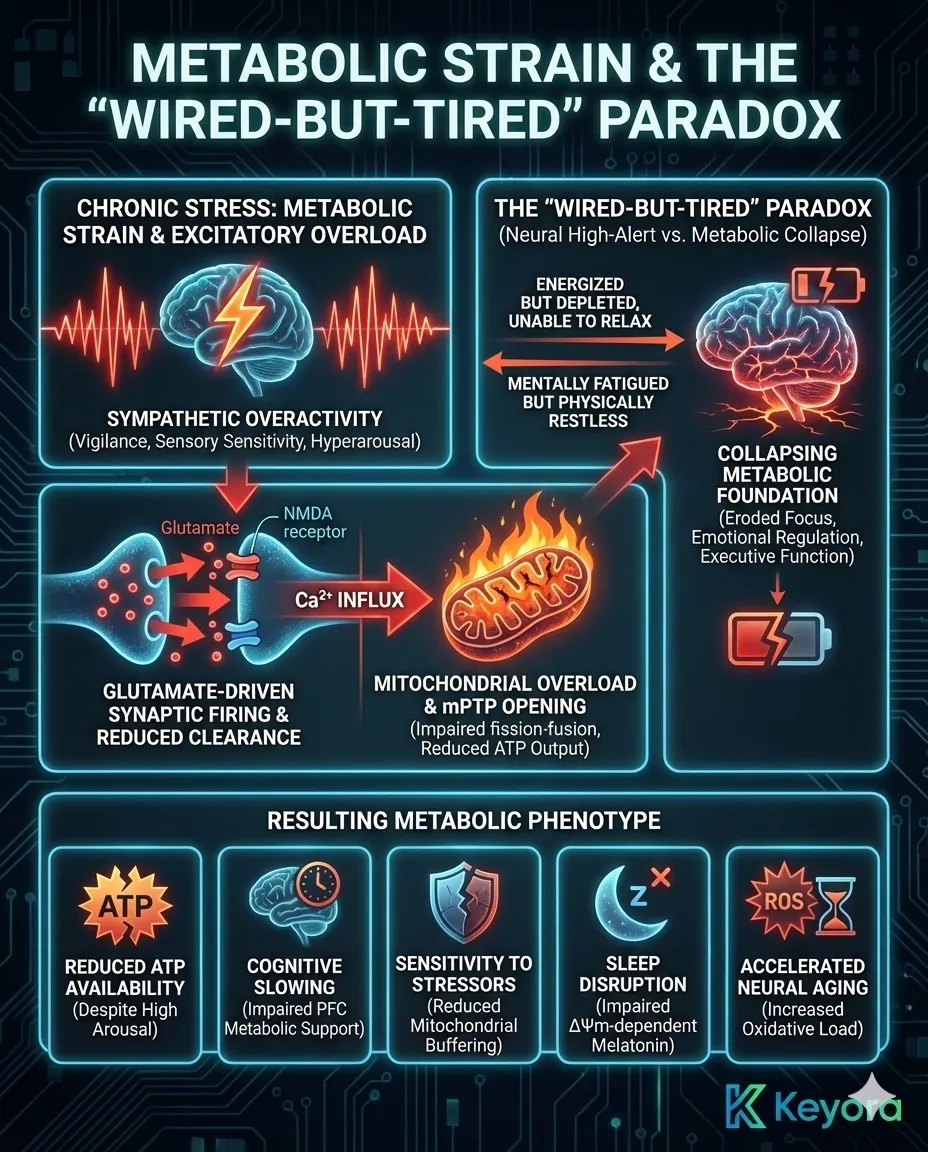
As metabolic strain accumulates, the brain shifts into a paradoxical operating mode: the sympathetic system remains overactive – maintaining vigilance, sensory sensitivity, and hyperarousal – while the energy systems needed for sustained focus, emotional regulation, and executive function erode.
This mismatch explains why individuals under chronic stress often feel “energized but depleted,” “unable to relax,” and “mentally fatigued but physically restless.”
Their neural systems are locked in a high-alert state, but the metabolic foundation needed to support that state is collapsing.
Excitatory neurotransmission further exacerbates this energetic mismatch. Glutamate-driven synaptic firing demands large amounts of ATP to maintain ionic gradients via Na⁺/K⁺-ATPase pumps. In stressed states, glutamate clearance is reduced, leading to prolonged NMDA receptor activation and higher calcium influx.
Excess intracellular calcium disrupts mitochondrial dynamics, impairs fission–fusion balance, and triggers opening of the mitochondrial permeability transition pore (mPTP), further reducing ATP output. This calcium-mitochondria overload cycle accelerates neurofatigue and cognitive decline.
The resulting metabolic phenotype includes:
• Reduced ATP availability despite high perceived arousal
• Cognitive slowing due to impaired PFC metabolic support
• Sensitivity to stressors because of reduced mitochondrial buffering capacity
• Sleep disruption as ΔΨm-dependent melatonin synthesis becomes impaired
• Increased oxidative load leading to accelerated neural aging
This phenotype aligns precisely with clinical observations in burnout, chronic stress syndromes, academic overload, professional exhaustion, and early-stage stress-related depressive states – all of which share mitochondrial inefficiency as a mechanistic core.
The “wired-but-tired” state is therefore a metabolic paradox produced by neurobiological imbalance.
The brain cannot downregulate arousal circuits due to limbic dominance and HPA dysregulation, yet it cannot maintain the metabolic output required to support the elevated neural firing.
It is a system trapped in high-alert mode without the energy to sustain it.
Understanding this mitochondrial collapse is crucial for appreciating why adaptogenic and metabolic interventions – such as Ashwagandha – play a uniquely restorative role.
Through modulation of cortisol rhythms, reduction of inflammatory signaling, stabilization of mitochondrial membranes, enhancement of antioxidant capacity, and support for ATP production, Ashwagandha targets the very nodes of this energetic imbalance.
This prepares the conceptual foundation for Episode 2, where the HPA axis will be analyzed as the primary upstream driver of mitochondrial strain.
– Chronic stress overloads brain mitochondria: hyperarousal + high metabolic demand + inflammation.
– Mistimed or elevated cortisol damages ETC complexes I & III, lowers ΔΨm, increases ROS.
– Cytokines (TNF-α, IL-1β) + iNOS → excess NO → peroxynitrite → mtDNA and membrane damage.
– Result: reduced ATP production, impaired neuronal function, particularly in PFC and hippocampus.
– Glutamate accumulation → calcium influx → mitochondrial permeability transition → worsening ATP loss.
– Produces “wired-but-tired” paradox: hyperarousal with metabolic failure.
– Clinical consequences: cognitive fatigue, emotional instability, insomnia, stress hypersensitivity.
– Sets biological stage for adaptogenic interventions (e.g., Ashwagandha).
Section VI
Integrating the Collapse: The Systems Failure Model of Chronic Stress
Chronic stress does not damage the brain and body through a single pathway. Instead, it produces a multi-system breakdown through tightly interlocked biological loops that reinforce each other.
What begins as an adaptive stress response gradually evolves into a pathological network state – one in which cortisol rhythms collapse, inflammation becomes self-sustaining, cognitive regulation fails, and mitochondrial energy systems break down.
Section VI integrates these processes into a unified systems-failure model that describes how stress transforms from an event into a persistent biological condition.
At the top of the cascade lies the collapse of the cortisol rhythm described in Section II.
Rather than delivering precise circadian pulses, cortisol becomes erratic – flattened, inverted, or chronically elevated at the wrong times. This timing breakdown is not just hormonal noise; it acts as the first systemic destabilizer.
Cortisol’s rhythmic pulses normally synchronize the immune system, metabolic cycles, neural arousal patterns, and sleep–wake architecture.
When rhythmicity collapses, downstream systems lose temporal coordination, entering asynchronous operation. This desynchronization is the structural foundation of stress-related disorders.
On this foundation, neuro-inflammatory coupling (Section III) becomes pathological. Without cortisol’s anti-inflammatory gating, immune cells remain in an activated state, producing IL-6, TNF-α, IL-1β, and other cytokines in a continuous loop.
These cytokines then act on the brain, priming microglia, amplifying excitotoxic responses, and further disrupting regulatory circuits.
The loss of vagal anti-inflammatory tone and the dominance of sympathetic signaling lock inflammation into a chronic state. This inflammatory load not only affects mood and cognition but reshapes neural architecture and metabolic function.
Simultaneously, the prefrontal–limbic imbalance outlined in Section IV becomes the central neural manifestation of chronic stress. The prefrontal cortex (PFC) weakens due to cortisol toxicity, glutamatergic overload, and inflammatory signaling. Its ability to regulate threat responses, manage attention, and modulate emotion diminishes. As a result, the amygdala becomes hyperactive, driving excessive anxiety, hypervigilance, emotional reactivity, and intrusive fear learning. This limbic dominance amplifies HPA activation, increasing cortisol output, which in turn further damages the PFC—closing a loop of escalating dysregulation.
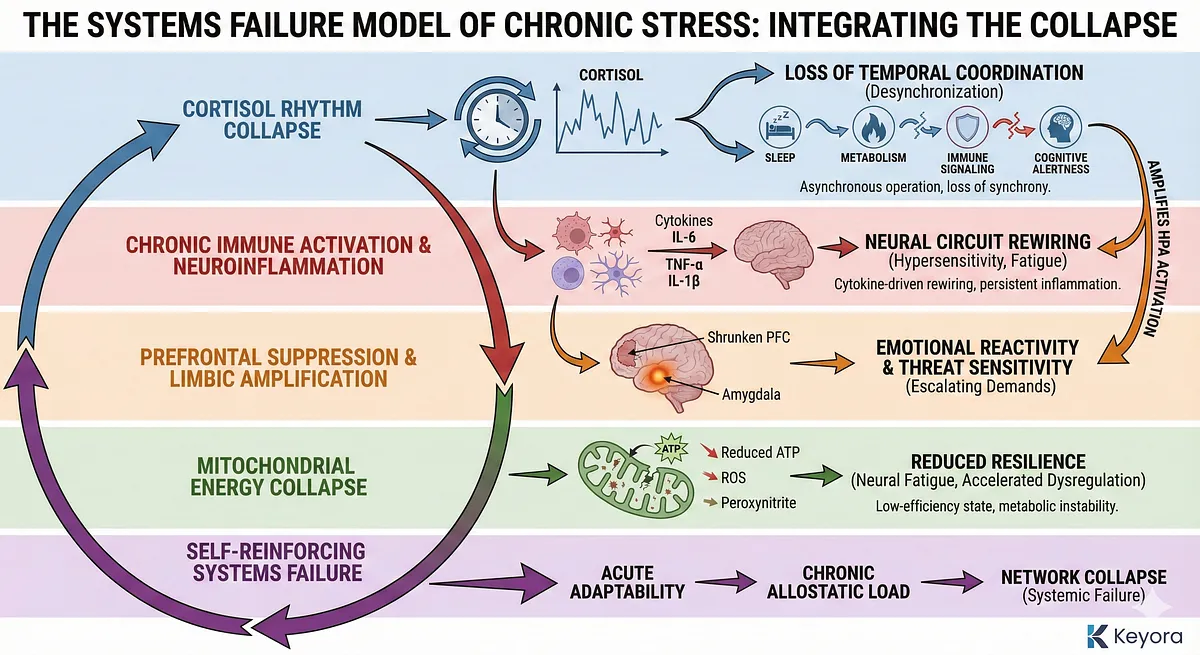
The metabolic layer adds another dimension to this collapse. Section V demonstrated how mitochondrial overload introduces energetic instability.
Neurons require enormous ATP supplies to maintain synaptic firing, inhibitory balance, ion gradients, and network plasticity.
Under chronic stress, mitochondria face simultaneous threats: oxidative damage, disrupted electron transport chain function, impaired membrane potential (ΔΨm), and toxic cytokine–NO–peroxynitrite interactions. As energy systems fail, neurons shift into a low-efficiency state, producing the “wired-but-tired” phenotype – hyperaroused yet depleted.
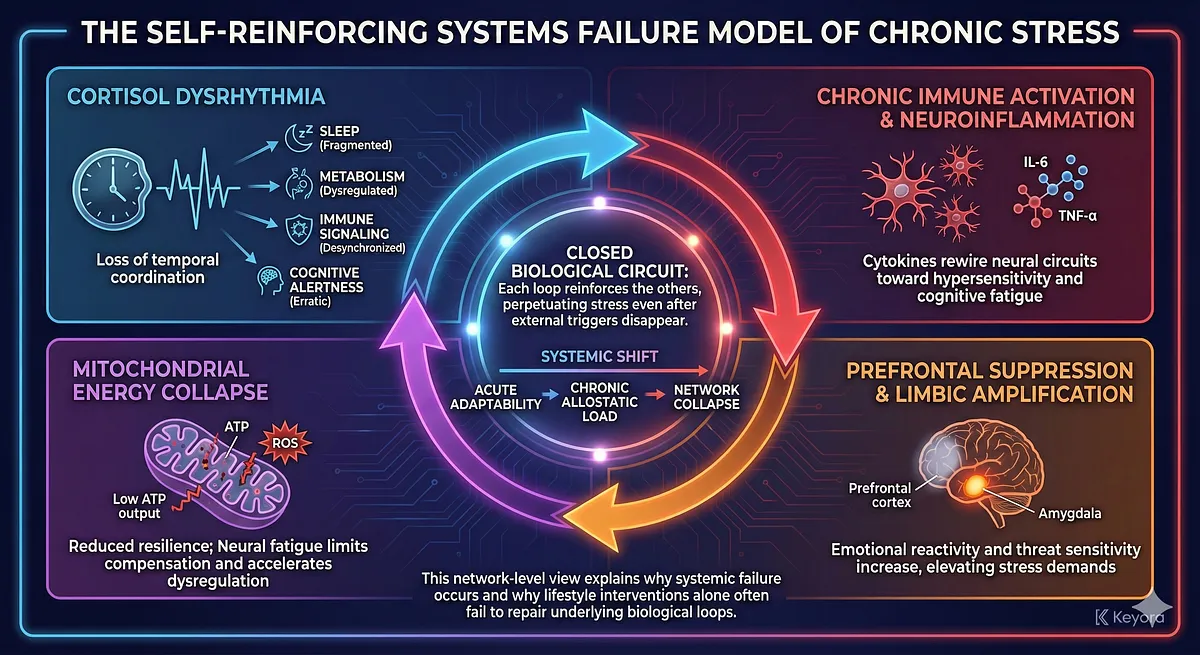
When combined, these layers form a self-reinforcing systems failure model:
- Cortisol dysrhythmia → loss of temporal coordination
Sleep, metabolism, immune signaling, and cognitive alertness lose synchrony. - Chronic immune activation → neuroinflammation
Cytokines rewire neural circuits toward hypersensitivity and cognitive fatigue. - Prefrontal suppression → limbic amplification
Emotional reactivity and threat sensitivity increase, elevating stress demands. - Mitochondrial energy collapse → reduced resilience
Neural fatigue limits compensation and accelerates dysregulation. - Each loop reinforces the others
Creating a closed biological circuit that perpetuates stress even after external triggers disappear.
The system thus moves from acute adaptability → chronic allostatic load → network collapse.
This network-level view explains why individuals under chronic stress experience symptoms that appear psychological, metabolic, cognitive, immune-related, and emotional – because the failure is not localized but systemic.
It also explains why lifestyle interventions alone often fail: they attempt to modify behavior without repairing the underlying biological loops that perpetuate the stress state.
This systems-failure model sets the conceptual stage for the entire Ashwagandha series.
To correct chronic stress, interventions must act not on a single mechanism but on multiple nodes within the network: cortisol rhythmicity, inflammation, neurotransmission, mitochondrial function, and neural structural integrity.
In Episode 2, we will begin dissecting the HPA axis as the master upstream driver of these failures, and explain how Ashwagandha’s molecular actions target not one but multiple layers of this collapsing system.
– Chronic stress causes multi-system failure through interlocking biological loops.
– First destabilizer: cortisol rhythm collapse → loss of circadian coordination.
– Immune layer: chronic cytokine activation (IL-6, TNF-α, IL-1β) → microglial priming → neuroinflammation.
– Neural layer: weakened PFC + hyperactive amygdala → emotional reactivity and impaired regulation.
– Metabolic layer: mitochondrial overload → ROS, ΔΨm loss, ATP insufficiency → “wired-but-tired.”
– These layers form a self-reinforcing circuit: stress persists even without external triggers.
– System moves from acute stress → chronic allostatic load → network collapse.
– Sets stage for multi-target interventions such as Ashwagandha.
Section VII
Why This Multi-System Breakdown Sets the Stage for Ashwagandha
The systems-failure model described in Sections I–VI reveals a stress state that is not psychological, not merely hormonal, and not simply emotional – it is multi-layered biological collapse.
The disruption spans cortisol rhythmicity, immune signaling, neurotransmission, energy metabolism, and neural architecture. Crucially, these components do not fail independently; they fail as an integrated network with self-reinforcing loops.
This is precisely why traditional single-target interventions – sleep hygiene, meditation, or isolated nutritional support – often cannot fully restore stability. Chronic stress is not a surface-level problem; it is a deep systems problem. And systems problems require systems-level interventions. This is the conceptual platform on which Ashwagandha becomes uniquely relevant.
Ashwagandha is not a sedative, not a stimulant, and not a simple antioxidant.
It is a broad-spectrum adaptogen whose molecular actions simultaneously influence multiple nodes within the very loops that drive stress-related system collapse.
Its clinical value is not derived from a single mechanism but from its capacity to modulate several interconnected biological domains at once.
This multi-domain regulatory capacity aligns directly with the system breakdown illustrated earlier.
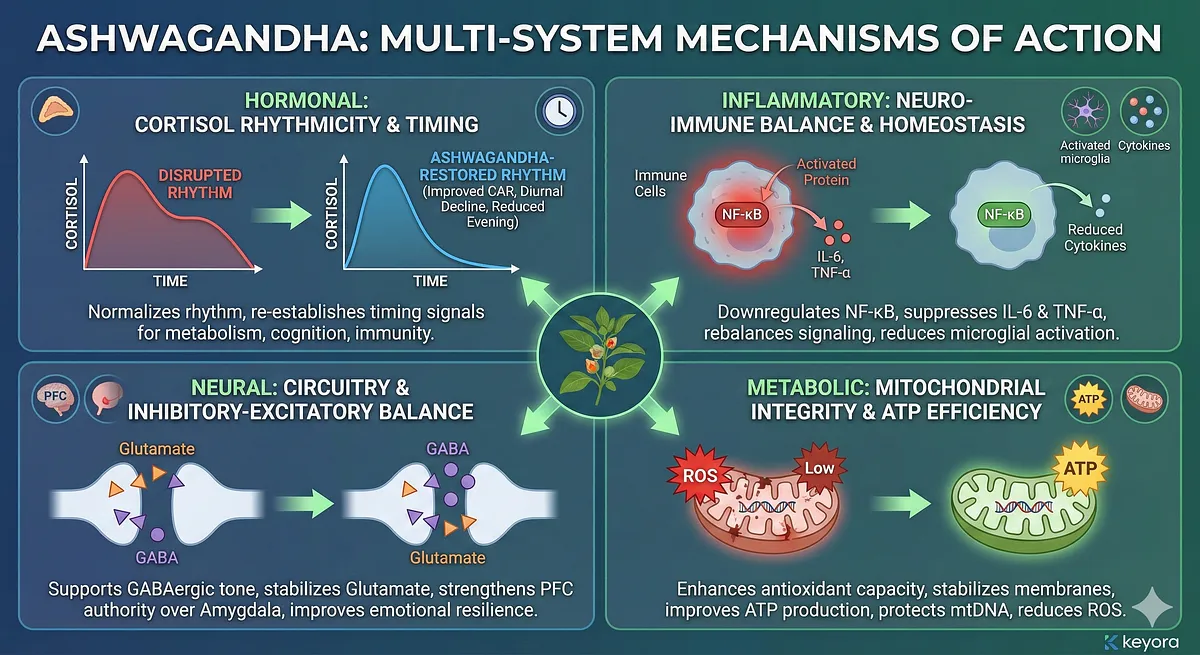
At the hormonal level, Ashwagandha is one of the few botanical compounds shown to normalize cortisol rhythmicity rather than merely lowering cortisol levels.
By improving the cortisol awakening response (CAR), restoring diurnal decline, and reducing inappropriate evening elevations, it re-establishes the timing signals that coordinate metabolism, cognition, and immunity.
This action directly addresses the initial destabilizer of the system – the collapse of biological periodicity.
On the inflammatory axis, Ashwagandha downregulates NF-κB activity, suppresses IL-6 and TNF-α signaling, and reduces microglial activation. R
ather than acting as a broad immunosuppressant – which would be maladaptive – it rebalances immune signaling toward homeostasis.
This counters the chronic neuro-inflammatory amplification loop described in Section III and interrupts the cytokine-driven remodeling of neural circuits.
On the neural circuitry level, Ashwagandha supports GABAergic tone, stabilizes glutamatergic activity, and improves the inhibitory–excitatory balance necessary for prefrontal cortex function. This provides a direct counter-force to the limbic dominance described in Section IV by strengthening the PFC’s regulatory authority over amygdala-driven responses. The result is improved emotional resilience, reduced hyperreactivity, and enhanced cognitive control.
Within the metabolic and mitochondrial domain, Ashwagandha enhances antioxidant capacity, stabilizes mitochondrial membranes, and improves ATP production efficiency.
It reduces ROS burden, protects mitochondrial DNA, and supports complex I activity – mechanisms directly relevant to the “wired-but-tired” energetic phenotype from Section V.
By improving neuronal energy availability, Ashwagandha enhances the brain’s capacity to recover from chronic overload and re-establish functional integrity.
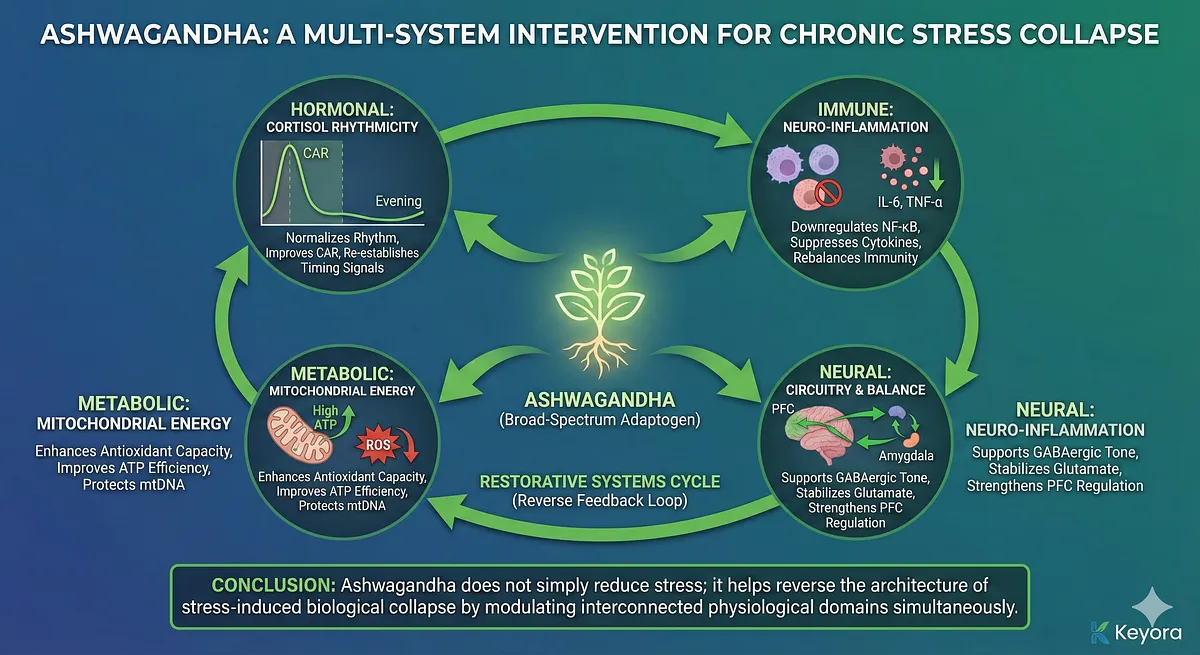
Critically, these effects do not operate in isolation. Restoring cortisol rhythmicity makes immune modulation more effective; reducing inflammation improves mitochondrial function; improving mitochondrial efficiency enhances prefrontal regulation; strengthening PFC networks reduces HPA overactivation. Each beneficial action reinforces others, creating a reverse feedback loop – a restorative systems cycle instead of a degenerative one.
In other words, Ashwagandha does not simply reduce stress; it helps reverse the architecture of stress-induced biological collapse.
This is why Ashwagandha is a cornerstone of the Keyora Nutritional Neurology framework. It is not positioned as a universal cure, nor as a superficial wellness product, but as a mechanistically grounded intervention for a deeply interconnected physiological problem.
Chronic stress is a systems disorder, and Ashwagandha is one of the rare nutraceutical compounds capable of addressing the system as a system.
Episode 1 has established the mechanistic landscape of stress collapse.
In Episode 2, we will begin mapping the HPA axis – the master regulator of stress physiology – and demonstrate how Ashwagandha interacts with the axis at molecular, endocrine, neuronal, and behavioral levels to restore resilience and biological coherence.
– Chronic stress causes multi-system breakdown: cortisol rhythm, inflammation, neurotransmission, energy, and neural architecture.
– Single-target interventions often fail because stress dysregulation is network-based, not linear.
– Ashwagandha functions as a multi-domain adaptogen affecting several stress nodes simultaneously.
– Hormonal layer: normalizes cortisol rhythm (CAR, diurnal decline, evening suppression).
– Immune layer: downregulates NF-κB, IL-6, TNF-α; reduces microglial activation.
– Neural layer: supports GABA tone, stabilizes glutamate, strengthens PFC regulation of limbic circuits.
– Mitochondrial layer: enhances ATP efficiency, reduces ROS, protects mtDNA.
– Multi-target actions form a restorative feedback cycle, countering degenerative stress loops.
– Establishes Ashwagandha as a systems-level intervention for a systems-level disorder.
– Prepares for Episode 2: mechanistic deep-dive into the HPA axis.
By Keyora Research Notes Series
This article contributes to Keyora’s ongoing scientific documentation series, which systematically outlines the conceptual foundations, mechanistic pathways, and empirical evidence informing our research and development approach.
ORCID: 0009–0007–5798–1996